Thalassemie, frequent bij personen met een migratieachtergrond
27-02-2024
Rode bloedcellen bevatten grote hoeveelheden hemoglobine (Hb), met als belangrijkste functie het zuurstoftransport van longen naar weefsels. Hb is steeds opgebouwd uit 4 eiwitketens (globine subunits) die elk één heemgroep bevatten (de zuurstofbindende molecule). Bij de volwassene vinden we voor 95-97% HbA1 (samengesteld uit 2 alfa-ketens en 2 bèta-ketens) en een kleine hoeveelheid HbA2 (2-3%) (waarbij de 2 bèta-ketens vervangen worden door 2 delta-ketens). Bij volwassenen (en steeds bij jonge kinderen) komt nog een kleine hoeveelheid foetaal hemoglobine (HbF) voor (gamma-ketens i.p.v. bèta-ketens) (Figuur 1, rechts).
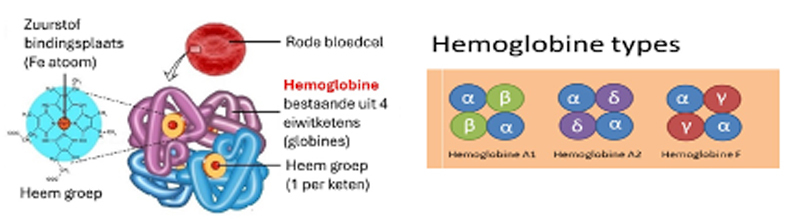
Figuur 1. Links: structuur hemoglobine A1 molecule, een tetrameer met 2x2 subunits die elk één heemgroep hebben. Rechts: opbouw van de verschillende hemoglobine types die kunnen gevonden worden bij een gezonde volwassen persoon.
Thalassemieën zijn overerfbare aandoeningen waarbij één of meerdere genen, die coderen voor de alfa- of bèta-ketens van het Hb, afwijkingen vertonen. Het belangrijkste effect is een verminderde productie van de alfa- of bèta-globineketens. Dit heeft als gevolg een verminderde aanmaak van goed functionerend Hb, met (mogelijks) een microcytaire, hypochrome anemie als consequentie.
Alfa-thalassemie (verminderde productie α-ketens) komt vooral voor in Zuidoost-Azië en China. Bèta-thalassemie (verminderde productie β-ketens) wordt vooral gezien rond het Middellandse zeegebied (Italië, Griekenland, Turkije), in het Midden-Oosten, en ook Zuidoost-Azië en delen van Afrika.
Als autosomale recessieve overerfbare aandoening kunnen we een onderscheid maken tussen dragers (heterozygoten) en patiënten (homozygoten). Voor bèta-thalassemie zijn er twee genen. Voor alfa zijn er 4 genen en is de complexiteit groter (er kunnen 1, 2, 3 of 4 genen aangetast zijn). Van belang is dat 2 (asymptomatische) dragers een risicopaar vormen, waarbij er 25% kan is op een kind met een homozygote (ernstige) afwijking.
Kliniek
Alfa-thalassemie
- 1 of 2 aangetaste genen: mogelijks milde asymptomatische microcytaire anemie.
- Bij aantasting van 3 alfa-globine genen: klinische presentatie variërend van microcytaire anemie tot ernstig beeld met transfusie-afhankelijkheid.
- Als alle 4 alfa-globine genen zijn uitgeschakeld: ernstig ziektebeeld met hydrops foetalis en overlijden voor of rondom geboorte.
Bèta-thalassemie
- Bèta-thalassemie minor (heterozygote drager) : (al dan niet) lichte microcytaire anemie, in principe geen klachten.
- Bèta-thalassemie intermedia: tussenvorm (genetisch hetero-of homozygoot): kliniek matig tot ernstig, afhankelijk van de hoeveelheid bèta-globineketens die nog geproduceerd wordt.
- Bèta-thalassemie major (homozygoot)
- Progressieve, ernstige anemie. Omdat een pasgeborene aanvankelijk nog gamma-globineketens (HbF) heeft, treedt de anemie progressief op vanaf de leeftijd van 6 maanden.
- Bleek, icterisch, kortademigheid, slechte groei, galstenen (ten gevolge van hemolyse); na een tijd botvervormingen (‘chipmunk face’) en hepatosplenomegalie ten gevolge van extramedullaire hematopoëse.
- Transfusie-afhankelijk, en gevaar op ijzerstapeling door vele transfusies.
Diagnose
Het typische bloedbeeld van een thalassemie (drager) is als volgt (zie Figuur 2 voor een praktijkvoorbeeld): chronische microcytaire (MCV ↓), hypochrome (MCH ↓) anemie met polyglobulie (RBC ↑) bij normale ijzerstatus en afwezigheid van inflammatie (ferritine, sedimentatie en CRP alle normaal). Bij ernstigere gevallen zijn er bovendien tekens van hemolyse (haptoglobine ↓, indirect bilirubine ↑) en een beenmerg dat probeert de hemolyse te compenseren door een verhoogde aanmaak van RBC (reticulocyten ↑).
De diagnose van een bèta-thalassemie kan met een hemoglobine elektroforese worden bevestigd (hierbij worden de circulerende hemoglobine fracties gekwantificeerd; HbA2 zal gestegen zijn). Bij alfa thalassemie is het HbA2 dan weer gedaald op Hb electroforese, deze ziekte moet via genetisch onderzoek bevestigd worden. Voor aanvragen en interpretatie kan steeds met een klinisch bioloog overlegd worden.
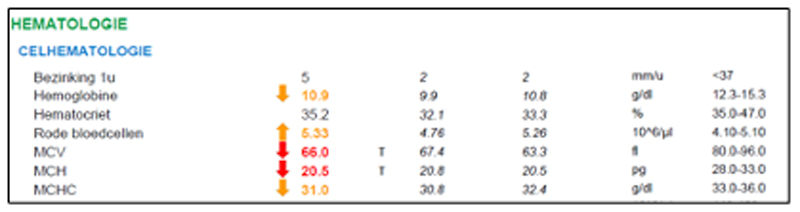
Figuur 2: een 42-jarige dame met migratieachtergrond vertoont sinds jaren een microcytaire, hypochrome anemie met polyglobulie bij normale ijzerstatus (DD ijzerdeficiëntie anemie) zonder inflammatie (DD anemie van de chronische ziekte). Hemoglobine elektroforese bevestigde de diagnose van bèta-thalassemie dragerschap (HbA2= 4.5%).
Rol van de huisarts
Vooral bij mensen met een migratieachtergrond (gebieden cf. supra) dient aan deze aandoeningen gedacht te worden, zeker bij zwangerschap(swens). Het is dan aanbevolen om ook de partner te testen en indien positief dient genetische counseling te gebeuren (verwijzing naar genetisch centrum). Dit om ernstige homozygote thalassemieën bij het nageslacht te vermijden.
Een algemene aanbeveling is ook om geen ijzersuppletie voor te schrijven bij personen met een migratieachtergrond zonder analyse van een hemoglobinopathie. Merk op dat naast thalassemieën, er nog andere hemoglobinopathieën zijn zoals sikkelcelanemie, HbE, HbC, etc. Deze worden ook opgespoord met de hemoglobine elektroforese.
Bij ernstige casussen (icterus, ernstige anemie) wordt verwijzing naar de hematoloog aanbevolen.