Thalassémie, fréquente chez les personnes issues de l'immigration
27-02-2024
Les globules rouges contiennent de grandes quantités d'hémoglobine (Hb), dont la principale fonction est de transporter l'oxygène des poumons vers les tissus. L'Hb est toujours composée de 4 chaînes protéiques (sous-unités de globine), chacune contenant un groupe hème (la molécule qui fixe l'oxygène). Chez l'adulte, on trouve 95 à 97 % d'HbA1 (composée de 2 chaînes alpha et de 2 chaînes bêta) et une petite quantité d'HbA2 (2 à 3 %) (où les 2 chaînes bêta sont remplacées par 2 chaînes delta). Chez les adultes (et toujours chez les jeunes enfants), une petite quantité d'hémoglobine fœtale (HbF) est encore présente (chaînes gamma au lieu de chaînes bêta) (figure 1, à droite).
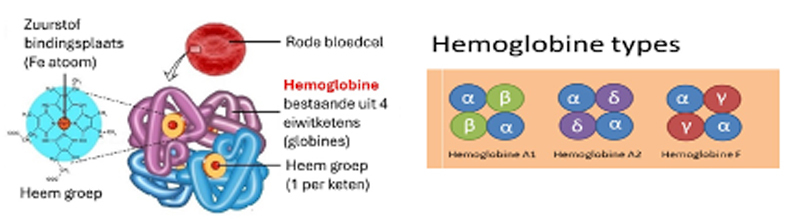
Figure 1. À gauche : structure de la molécule d'hémoglobine A1, un tétramère avec 2x2 sous-unités qui ont chacune un groupe hémique. À droite : structure des différents types d'hémoglobine que l'on peut trouver chez un adulte en bonne santé.
Les thalassémies sont des maladies héréditaires dans lesquelles un ou plusieurs gènes codant pour les chaînes alpha ou bêta de l'Hb présentent des anomalies. L'effet principal est une production réduite des chaînes alpha ou bêta de la globine. Il en résulte une production réduite d'Hb fonctionnelle, avec pour conséquence (éventuellement) une anémie microcytaire et hypochrome.
L'alpha-thalassémie (production réduite de chaînes α) est la plus fréquente en Asie du Sud-Est et en Chine. La bêta-thalassémie (production réduite de chaînes β) est surtout observée dans la région méditerranéenne (Italie, Grèce, Turquie), au Moyen-Orient, ainsi qu'en Asie du Sud-Est et dans certaines régions d'Afrique.
En tant que maladie héréditaire autosomique récessive, on distingue les porteurs (hétérozygotes) et les patients (homozygotes). Pour la bêta-thalassémie, il existe deux gènes. Pour la thalassémie alpha, il y a 4 gènes et la complexité est plus grande (1, 2, 3 ou 4 gènes peuvent être affectés). Il est important de noter que deux porteurs (asymptomatiques) forment une paire à haut risque, où il y a 25 % de chances d'avoir un enfant présentant une anomalie homozygote (grave).
Clinique
Alpha-thalassémie
- 1 ou 2 gènes affectés : possibilité d'une anémie microcytaire légère et asymptomatique.
- Si 3 gènes alpha-globine sont affectés : présentation clinique allant de l'anémie microcytaire à un tableau sévère avec dépendance transfusionnelle.
- Si les 4 gènes alpha-globine sont désactivés : tableau clinique grave avec anasarque fœtale et décès avant ou autour de la naissance.
Béta-thalassémie
- Béta-thalassémie minor (porteur hétérozygote) : (que ce soit ou non) anémie microcytaire légère, fondamentalement aucun symptôme.
- Béta-thalassémie intermedia: forme intermédiaire (génétiquement hétéro- ou homozygote) : clinique modérée à sévère, en fonction de la quantité de chaînes de bêta-globine encore produites.
- Béta-thalassémie major (homozygote)
- Anémie progressive et sévère. Comme le nouveau-né possède encore des chaînes de gamma-globine (HbF), l'anémie survient progressivement à partir de l'âge de 6 mois.
- Pâle, ictérique, essoufflé, faible croissance, calculs biliaires (dus à l'hémolyse) ; après un certain temps, déformations osseuses ("visage de chipmunk") et hépatosplénomégalie due à l'hématopoïèse extramédullaire.
- Dépendante des transfusions, et risque d'accumulation de fer en raison de nombreuses transfusions.
Diagnostic
L'image sanguine typique d'un porteur de thalassémie est la suivante (voir figure 2 pour un exemple pratique) : anémie chronique microcytaire (MCV ↓), hypochrome (MCH ↓) avec polyglobulie (RBC ↑) dans un statut en fer normal et en l'absence d'inflammation (ferritine, sédimentation et CRP tous normaux). Dans les cas les plus graves, on observe en plus des signes d'hémolyse (haptoglobine ↓, bilirubine indirecte ↑) et une moelle osseuse qui tente de compenser l'hémolyse par une production accrue de GR (réticulocytes ↑).
Le diagnostic de bêta-thalassémie peut être confirmé par une électrophorèse de l'hémoglobine (il s'agit de quantifier les fractions d'hémoglobine circulantes ; l'HbA2 sera augmentée). Dans le cas de l'alpha thalassémie, l'HbA2 diminue à nouveau à l'électrophorèse de l'Hb ; cette maladie doit être confirmée par un test génétique. Un biologiste clinique peut toujours être consulté pour les applications et l'interprétation.
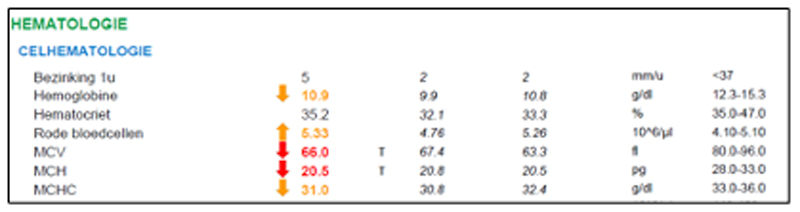
Figure 2 : Une femme de 42 ans d'origine migratoire présente depuis des années une anémie microcytaire et hypochrome avec polyglobulie dans un état ferrique normal (anémie par carence en fer DD) sans inflammation (anémie par maladie chronique DD). L'électrophorèse de l'hémoglobine a confirmé le diagnostic de porteur de bêta-thalassémie (HbA2= 4,5 %).
Rôle du médecin généraliste
Chez les personnes issues de l'immigration (cf. supra), ces conditions doivent être prises en compte, en particulier pendant la grossesse (swens). Il est alors recommandé de tester également le partenaire et, en cas de résultat positif, de procéder à un conseil génétique (orientation vers un centre génétique). Ceci afin d'éviter une thalassémie homozygote grave dans la descendance.
Il est également recommandé de ne pas prescrire de supplémentation en fer aux personnes issues de l'immigration sans analyse d'une hémoglobinopathie. Il convient de noter qu'outre les thalassémies, il existe d'autres hémoglobinopathies telles que la drépanocytose, l'HbE, l'HbC, etc. Celles-ci sont également détectées par électrophorèse de l'hémoglobine.
Dans les cas graves (ictère, anémie sévère), il est recommandé de consulter un hématologue.